|
何をもって有機伝導体の始まりとするか,というのはなかなか難しいものですが,有機物における伝導性の研究はまず,純物質を対象として行われました.例えばフタロシアニン系物質での伝導性の研究や,アントラセンの光伝導,井口先生によるより大きな縮合多環系炭化水素における光伝導の研究がこれに当たります[1].しかしながらこの段階では,伝導性があると言っても通常の金属と比べて108から1010程度も抵抗が高く,まだまだ導電性の物質とは呼べない状況でした. もう一つの問題は,これらのπ系分子のサンプルでの伝導は不純物の影響を受けやすく(この場合,不純物が混じると非常に抵抗が大きくなることが多い),測定者やサンプルによるばらつきが非常に大きい,というものでした.このため,見えている現象が本質なのか違うのかの区別がしにくく,研究の発展を妨げる大きな障害となっていました. こういった中,一つの大きな転機となったのが井口・赤松の両先生らによるペリレン-臭素電荷移動錯体の発見です.これはペリレンを溶かした溶媒中に臭素を加えることで得られる電荷移動錯体[2]ですが,電気伝導性が非常に高く,10-100 Scm-1と半金属であるゲルマニウムやテルルと同程度にまで達しています.これは後に,平面状のπ電子系化合物を酸化して伝導性を持たせる,という指針につながることとなります.  次の大きな発見は,DuPontによるTCNQの合成です.テフロンにより大成功を収めていたDuPontは,その類似物を作ろうと様々な試行錯誤を行っていました.テフロンのフッ素を同様に電子吸引性の高いシアノ基で置き換えTCNEを作り,これがうまくいかなかったためにさらに中央にベンゼン環を挿入してTCNQを開発したわけです.DuPontにとっては残念なことに,TCNEもTCNQも重合せずテフロン的な物質は得られませんでしたが,電子吸引基であるシアノ基を4つも含む構造から,ドナー性物質と組み合わせれば電荷移動を起こし,ペリレン-臭素のように高導電性が得られるのではないか,という観点で非常に多くの研究が行われました. その中でも大きな一歩となったのがNMP-TCNQです[3].この塩は室温で非常に高い伝導性(約200 Scm-1)を示すだけではなく,室温(以上)から200 Kまでの間で(半)金属状態が実現していました.(なお,DuPontの当初の思惑とは異なりますが,TCNQ錯塩は固体電解コンデンサなどに利用されています) 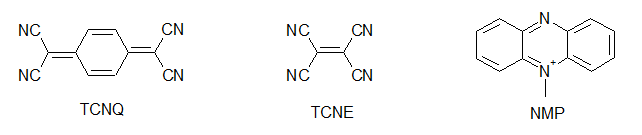 次に現れたのがTTF-TCNQです.これはそれまで用いられていたドナーとは大きく構造の異なる(といっても現在では有機導体のドナーはほぼこの系列になっていますが),硫黄を含むドナー分子であるTTFを用い,TCNQと組み合わせたものです.この結晶中では,TTFがδ+に,TCNQがδ-になるように電荷移動を起こし,それぞれの分子が作る1次元カラムが両方とも金属伝導を示す系となりました[4]. この物質では室温での伝導率が数百Scm>-1,低温ではさらに一桁程度上がるというように,それまでの有機物を大きく超える高導電性を示す系となりました. この物質は60 K付近で金属-絶縁体転移を起こし絶縁化しますが,当初は測定上のミスによりこの転移直前で抵抗が大きく減少すると報告され,超伝導が起きかかっているのだ,と大きな注目を集めました.ちょうど少し前にLittleが有機分子等で起き得る超伝導の新たな機構(転移温度が室温を達成できると思われた機構)を提案しており,有機導体こそが室温超伝導を目指せる系なのではないか,と多くの研究者が飛びつきました(無機物における高温超伝導体が現れる以前の話です).まあ,実際には測定ミスだったわけですが,これがきっかけで有機導体の研究者が一気に増えたという影響も与えています. 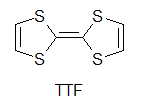 こうして,TTFの優秀さが知れ渡ると,これを少し変更した分子群を使って,低温まで金属になるような系の探索が行われ始めます.また,ドナーとアクセプタを混合するという化学的な酸化だけでなく,アニオンを支持電解質として含む溶液中でドナー分子を電気化学的に酸化して,中性ドナーと酸化されたドナーが同時に結晶中に取り込まれることで部分酸化されたドナーのカラムを含む結晶を作る(D+ + nD0 → (Dn+1)+),という現在まで続く導電性有機物の結晶を作る優れた手法も導入されるようになります.こういった部分酸化塩では,生じたホールが隣の分子に移動してもクーロン力による損がほとんど無いことが多く,金属伝導を含め良好な導電性を示し,かつ質の良い結晶が多数得られるようになりました. そんな中生み出されたのがTMTSFです.メチル基が付くことで溶解性やパッキングの良さが上がっていますし,硫黄よりも隣接分子との間で軌道の重なりを持ちやすいセレンを用いることで,平板状の分子が積み重なるカラム方向だけでなく,隣接カラムとの間でも電子の移動が起こりやすい(=二次元的な電子状態となりやすく,1次元不安定性に基づく低温での絶縁化が置きにくいと期待できる)ドナー分子です. Bechgaardはこの分子を用い,いわゆる"Bechgaard Salt" (TMTSF)2PF6を構築します.これはそのままでは低温で絶縁化してしまったのですが,彼はこれを物性物理の実験家であるJeromeのところに持ち込みます.1次元不安定性で絶縁化するならば,圧力をかけて結晶を縮め,横方向での電子の移動を容易にすれば金属化するのではないか,と考えたわけです. そうして圧を印可してみたところ,この結晶は見事,有機物で初の超伝導(Tc = 1.6 K)を示すこととなります[5].その後,PF6-と同じような(けれども少しサイズが違う)丸いアニオンであるBF4-,ClO4-,AsF6-,SbF6-を使って結晶中のドナー分子の配列を少しのばしたり縮めたりすることで,外部から圧力を加えたのと同様の効果を与えるという化学圧力を用いた研究などへと発展していきます.こういった化学圧力は,圧力や化学修飾によって絶縁相から少しずらし,金属相と絶縁相の境界に存在する(事が多い)超伝導相を狙う,という方針とともに典型的な研究の手法として確立されています. 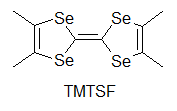 TTFの発展として,さらに外側に硫黄を含む環を付加したBEDT-TTF(通称ET)も生み出されます.こちらは斉藤軍治先生(いわゆるMr. BEDT-TTF)が精力的に物質開発を行い,この分子が非常に金属伝導や超伝導を示す物質を与えやすいことが明らかとなりました[6].これは一つには外側にSをつけることでπ系が広がり,分子内での電子間反発が減ることで導電性に有利に働くこと,また分子の横方向へも容易に重なりが生まれ二次元化しやすいこと,分子長軸方向に多少ずれてスタックしても(縦に長いπ系のおかげで)導電性カラム内での軌道の重なりが十分大きくなることなどに由来します. さらに横方向への接触を増やすために中央にSeを入れたBEDT-TSF(通称BETS)も大きな成功を収め,数多くの超伝導体を生み出すこととなりました[7]. 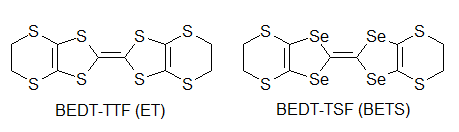 またこの頃から,アニオンとして磁性錯体を用いることで導電性のπ系と,磁性を担うd電子系が共存するような系の開発も行われるようになります.これはある種,無機物におけるs-d系を模倣しようというものとも言えるかも知れません.そういった中から,例えば磁性アニオンの存在により超伝導がサプレスされるλ-(BETS)2MCl4(非磁性のM = Gaでは超伝導になるが,磁性アニオンのM = Feの場合は同じ温度で絶縁化する)[8],反強磁性秩序と超伝導が共存するκ-(BETS)2FeBr4[9],強磁性と金属伝導が共存する(BEDT-TTF)3[MnCr(ox)3][10]が生み出されました.特にλ-(BETS)2FeCl4に関しては,磁性アニオンの存在によりサプレスされていた超伝導相が強磁場を印可することにより復活する(磁場誘起超伝導)といった現象も見つかっています[11]. 超伝導を目指すという側からは,これまでの流れとは逆にπ系を縮小して電子相関を増やす,という設計指針が生まれています.横方向の重なりを増やし次元性を向上し,π系を広げて分子内での電子相関を減らすというのは導電性を向上するには良い指針だったわけですが,超伝導というのは絶縁相と金属相との境に発生しているものですから,あまり金属相を安定化しすぎると単なる金属となってしまいます. そこで,分子内での電子間反発を増やし金属相を不安定化させる,という目的のもと,TTF骨格の一部を水素化してπ系を縮小したDHTTF(dihydro-TTF)を基本骨格とした分子群が開発されました. これらの分子を用いた結晶では,π系が縮小されたことにより金属相が不安定化し,いくつもの超伝導体を生み出しています[12]. 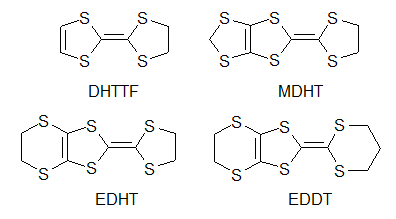
[1] H. Inokuchi, Bull. Chem. Soc. Jpn., 27 (1954) 22 |