アセチリドでπ-d系を
|
有機導体(伝導性のπ電子系)と磁性錯体(局在d電子系)を組み合わせたπ-d系は,磁場誘起超伝導や巨大磁気抵抗効果など,単に伝導と磁性が共存しているだけではなく,両者の間の相互作用による新たな複合物性が表れる興味深い系です.しかし残念ながら,多くの場合では電気伝導を担うπ電子と,磁性を担うd電子との間の相互作用が非常に弱く,おおざっぱに言って2J / kBの大きさで1から数ケルビン程度にしかなりません.この相互作用の弱さのため,面白い物性が現れるのは極低温に限られ,さらにしばしば他のもっと強い相互作用に阻害され表に現れない,などと言うことも生じます.従って,π-d相互作用を強くすることはこの分野の研究者が常に考えているテーマでもあります. このπ-d相互作用,なぜ弱いのでしょうか?実は弱い理由は単純で,伝導を担う電子と磁性を担う電子が異なる分子上に存在し,両者の間の距離が非常に離れている,というところが問題です.例えば酸化物系などの無機導電性磁性材料の場合,伝導を担う電子と磁性を担う電子は同じ原子上に存在し強い相互作用で結ばれています.ところが一般的なπ-d系の場合,導電性の分子と磁性分子では直接接触している原子間でも4 Å程度,分子の中心同士の距離で言えば10 Å以上も離れており,両者の相互作用は相当に弱くなってしまいます.
さて,磁性と伝導が異なる分子上に存在しているのが悪いのなら,両者を同じ分子に組み込んでしまえばいいじゃないか,というのは非常に素直な発想です.当然このような発想に基づいた分子設計はいくつも行われており,TTF骨格を遷移金属に配位させた例[1-5]や,導電性錯体が中心金属として磁性イオンを含む系であるフタロシアニンでの研究[6],遷移金属ではありませんが有機ラジカルをTTF骨格に結合させた系[7]などが存在しています. そんな中,今回私が作成した新規錯体[8-10]は,クロムアセチリドをベースとした以下の錯体です.
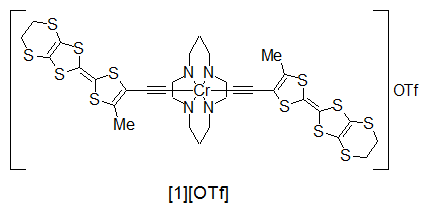
配位子としてはMeEDT-TTFにエチニル基を付けたものを用いています.Me基は合成の容易さと溶解度の向上のために導入,EDTの部分はπ系を拡張して隣接分子と相互作用しやすくするためです.中心金属はCr3+を用いており,S = 3/2と比較的大きなスピンを持ちます.このCrCyclamを中心に据えたアセチリド錯体は非常に安定であることが知られており,様々なアセチリド錯体を今後開発する際にも便利であると考え採用しました.
今回は新規錯体と言うこともあり,まず酸化に耐えられるのかを調べるため,CVで電気化学的な特性を測定しました.その結果が以下のグラフです.
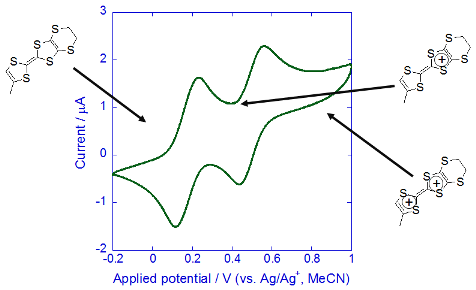
TTF骨格に由来する2段階の酸化還元が見えており,Cr3+の部分はそのままに,TTF骨格が選択的に酸化されていることがわかります.またきれいに可逆的な酸化還元を示していることから,酸化されても錯体自体は安定に存在していることも示唆されます.なお,錯体1分子につきTTF骨格は2つ配位していますが,それらの酸化還元は分離して見えてはおらず同じ位置で酸化されています.これは二つの配位子の距離が遠く,両者の間に働くクーロン力がかなり小さいためだと考えています. 酸化に耐えることがわかったので,いよいよ導電性磁性結晶を目指して電解合成の開始です.有機導体でよく用いられる以下のセッティングに従い,こちらの定電流電源で電解を行います.アニオンとしてはTBA[ClO4]やTBA[BF4]などの1価の4面体アニオンを用いました.

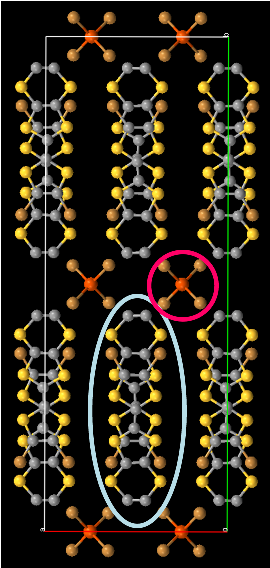
数週間もすれば電極表面に黒色の小さな結晶(運が良ければ長い針状結晶の場合もあり)が析出してきます.得られた結晶は基本的にアニオンの違いによらず同じ結晶構造で,以下のような構造でした.なお,CIFファイルはこちら.
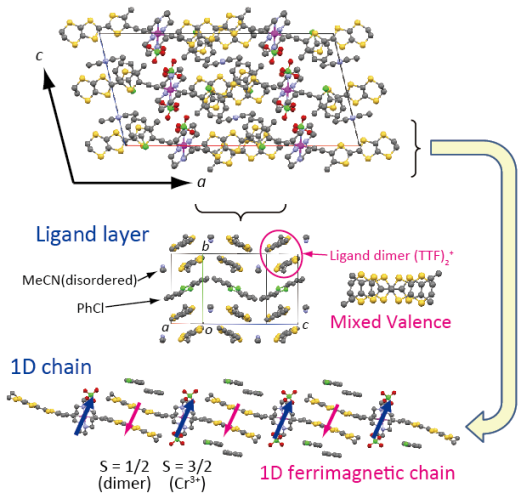
結晶の組成は[CrCyclam(C≡CMeEDT)2][Anion]2(PhCl)2(MeCN)で,C2/c の空間群.結晶学的に独立な要素はアセチリド錯体が半分にアニオン1つ,溶媒分子がそれぞれ1つです(MeCNはdisorderあり).アセチリド錯体1分子につきアニオンが2分子存在しているので,[CrCyclam(C≡CMeEDT)2]2+という酸化状態(もとの錯体+1価)になります.CVの結果から+1価の酸化はTTF骨格で起こっていることが推測されますが,錯体1つあたりに存在する配位子は2つでしかも結晶学的には等価.どうなっているのかと言えば,隣接する錯体との間でTTF二量体を組み,+1価の電荷(とそれに付随するS = 1/2のスピン)が二つのTTF骨格,つまり隣接分子間で非局在化しています. この結果,Cr3+-C≡C-(TTF)2+-C≡C-Cr3+……という1次元フェリ鎖が形成され,結晶の基本構造となっています. フェリ鎖間ではTTF二量体間にわずかな接触があり,ここを介して反強磁性的に相互作用していることが予想されます.なお,上図であるフェリ鎖と,c軸方向に並んでいる隣のフェリ鎖とは二回軸で結ばれる関係となっており,両者の間に反転中心は存在していません(これが後に述べる磁性の原因となる).TTF二量体はほぼ孤立しているため,残念ながら現段階では導電性は期待できません.導電性の共存は今後の課題です. 磁性は高温域ではフェリ鎖に由来する磁性を示し,フィッティングから鎖内相互作用は2J / kB = -30 K程度とかなり強い相互作用が働いていることが確認出来ました.この相互作用はTTF二量体とCr3+との間,つまりπ-d間に働く働く相互作用となります.これが強いということは,π系とd系をエチニル基で繋ぐというアセチリド構造が,強いπ-d系を今後構築していく上で有望である事を示唆しています.
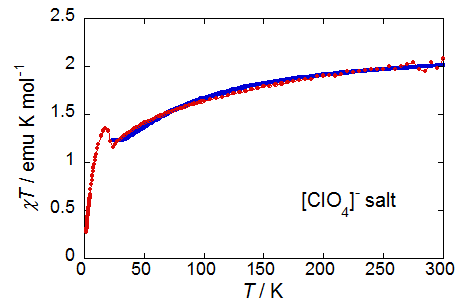
23 Kあたりに見える小さな飛びは磁気転移となり,低温ではc軸方向(ほぼTTF骨格のside-by-side方向)にスピンフロップが,これと直交する方向(結晶外形が棒状のため,ab面内のどの方向かは不明)に小さな自発磁化が現れます.
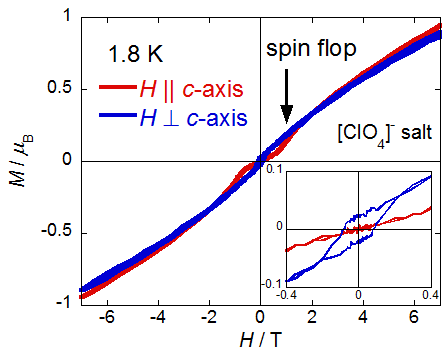
容易軸と自発軸の方向が直交していること,現れる自発磁化が小さいことから,この結晶は傾角反強磁性による弱強磁性体であることがわかります.この弱強磁性の起源に関しては,アニオンを[BF4]-,[ClO4]-,[ReO4]-と大きくするに従って
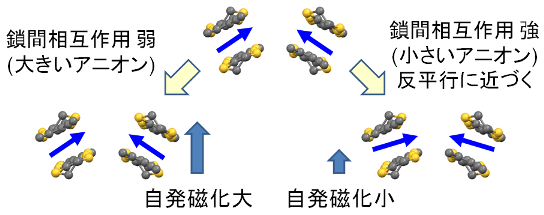
まず,スピンはTTF骨格に由来する異方性によりTTFのside-by-side方向に向きたがると考えます.ここで鎖間相互作用が弱いと,(転移温度は低くなるものの)この異方性が生き残ってきて隣接するフェリ鎖のスピン成分が打ち消されず弱強磁性を示します.一方,鎖間相互作用が強くなると,(転移温度は高くなるものの)スピンは強い反強磁性相互作用により反平行に近づき,自発磁化は小さくなるわけです. まとめると,今回のこの研究は
[1] D. Lorcy et al., Coord. Chem. Rev., 2009 253, 1398. |